The docking process involves two basic steps: prediction of the ligand conformation as well as its position and orientation within these sites (usually referred to as pose) and assessment of the binding affinity.
What is molecular docking and how it is performed?
Molecular docking is the study of how two or more molecular structures (e.g., drug and enzyme or protein) fit together [50]. In a simple definition, docking is a molecular modeling technique that is used to predict how a protein (enzyme) interacts with small molecules (ligands).
How do you do protein ligand docking?
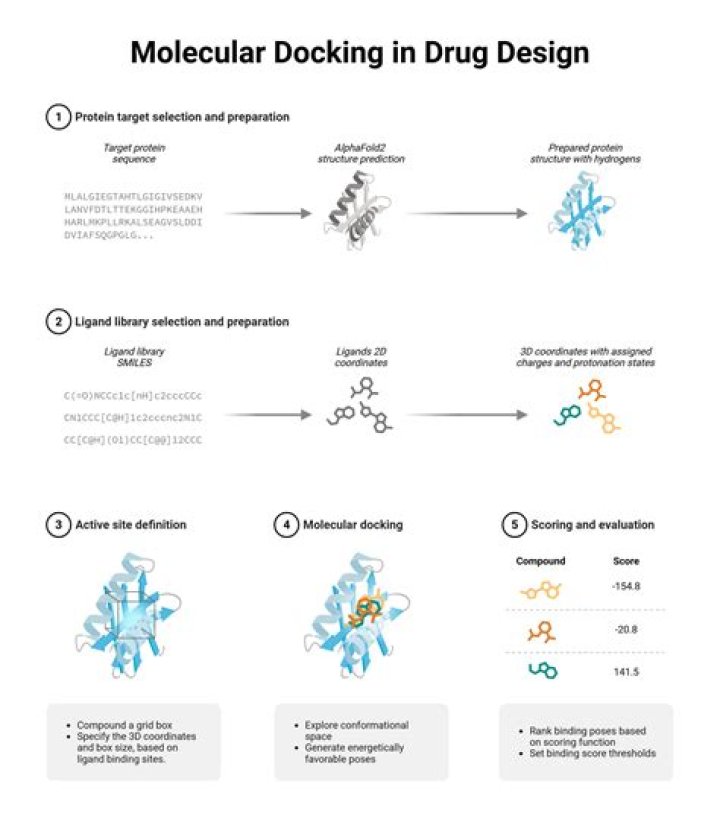
Following, we will describe the four-step procedure adopted in this study to perform the molecular docking.
- 3.1. Target selection.
- 3.2. Ligand selection and preparation.
- 3.3. Docking.
- 3.4. Evaluating docking results.
- 3.5. Docking software description.
What is the difference between Autodock and Autodock Vina?
The major difference is the scoring function. In Autodock 4 the scoring function is based on the AMBER force field and in Autodock Vina it is a hybrid scoring function (empirical + knowledge-based) based on the X-Score function with some different parameters which is not published at the moment.
How do I use Autodock in Windows?
The simplest way on windows 10 is going to the directory where autodock.exe is and open a terminal there (on windows 10 you can go to the upper left panel in “File” and then “Open WindowPowerShell). Automatically, a new terminal will appear in that specific direction. Then, type . \autodock.exe -h and it should work…
How do you dock an Autodock?
- 2.1 Preparation of Target.pdbqt file. Open File. Read Molecule.
- 2.2 Preparation of Ligand.pdbqt file. Open Ligand. Click Input.
- 2.3 Preparation of Grid Parameter File (a.gpf) Open Grid. Click Set Map Types.
- 2.4 Preparation of Docking Parameter File (a.dpf) Open Docking. Click Macromolecules.




